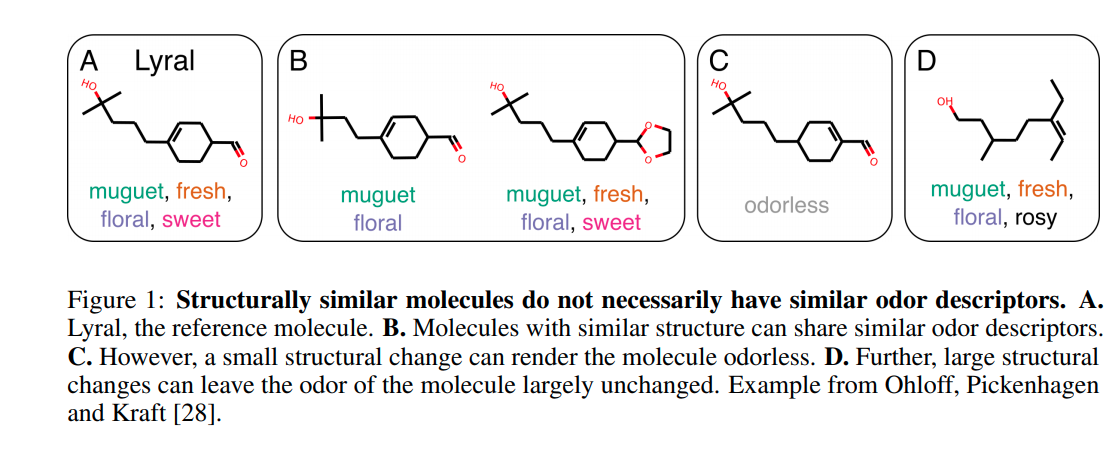
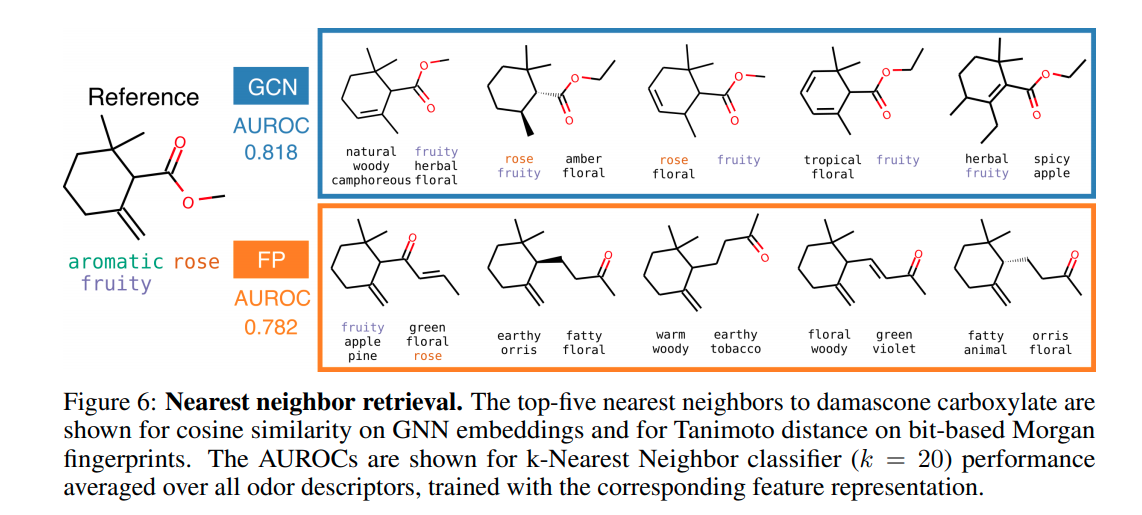
especially in figure 1 “Structurally similar molecules do not necessarily have similar odor descriptors.”
why is this so? is it because 3d information is not used (molecule are essentially 3-dimensional)
i have develop a basic graph CNN and now thinking of how to improve results. If structure cannot tells the odor, what else do?
Question1: The essence about ordor comes from molecule and protein interaction, it’s very complicated. Think about the key and lock, small change in key would make it not working.
Question2: A paper about it. Is It Possible to Predict the Odor of a Molecule on the Basis of its Structure?, I did not find an answer yet.
There is two importants information that you need to take into account.
Lot of the olfactive molecules are based or originally discovered in nature. Generally Nature is very economic so a little molecular modification can have a huge difference in term of Odor. This is basically due to the high interaction of a given molecule to a serie of olfactive receptors. In over words, this is not linear and really related to ligand protein interaction.
Two things that may help you:
when we know totally 3D of a molecule. it’s written in the SMILES. Using RDKit tool you can have a canonical (unique) representation of this molecule, E/Z represent (encoded by those charaters /,) is very important in the 3D, another one called R/S is also represented in the SMILES and can be encoded in GCN. If, we can produce / purify this particular molecule and smell it. Then you will have one (but human response can vary time to time) olfactive sentence to describe it.
when you cannot purify / separate / create a pure potentially chiral molecule. You may have in the best situation (if only one chirality center) two molecules in a mixture generally 50/50. in this conditions, you have the overlap of two olfactive descriptions (it’s not really linear cause the power of the two isomers can be also different!).
As you can see, we have like in pharmaceutical challenges and chemical databases a part of the molecules that are 3D unique and this part vary based on the target. Finally, in Pharma, between 30-50% of drugs are pure 3D molecules.
Also relevant to the paper above, the “DREAM Olfaction Prediction Challenge” is a good starter,
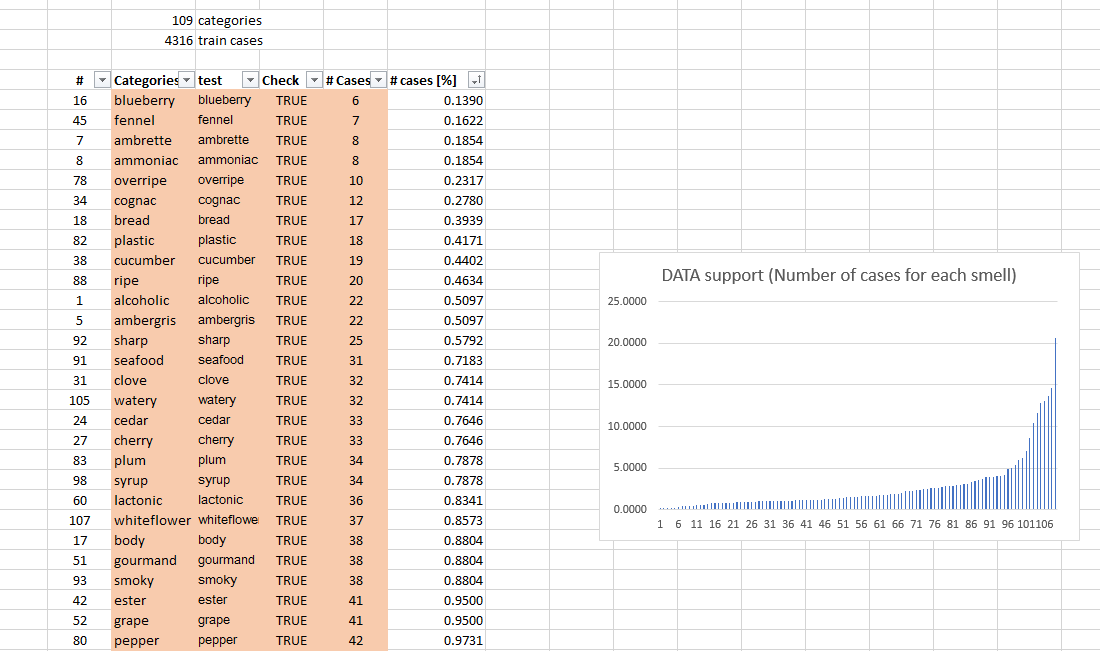
while NLP and SMILES decomposition may potentially work, using 2D and 3D chemical descriptors, or 3D/4D COMFA/COMSA or HQSAR descriptor approaches are probably the best. Software is linked below. The dataset of course has problems with only 13 smells having more than 5% cases and the rest of 93 cases having less than 5% support in the training set, there will be no stable predictions, unless the holdout set is maybe 10,000 compounds.
Code for traditional descriptor approaches (R/Python examples)
Some additional explanation
Free online 2D and 3D descriptor calculation website, ChemDes allows users to compute 3679 molecular descriptors from several open source http://www.scbdd.com/chemdes/
One challenge of course will be the 3D conformer generation, from SMILES, without changing chirality. If there is none, then which to pick, because in principle we don’t know that, as discussed before when we have racemic mixtures (R/S).
It would be interesting to ask what to do in such a case, the ML algorithm will potentially determine the smell of a racemic mixture, but for a correct chemical annotation you still would need to have the enantiomers (R/S) separated or you would have to pick on (if there are any). So there is lots of ambiguity here, probably one reason why its a challenge.
RDKit also provide a nice and fast 3D generator, thanks to the community (https://pubs.acs.org/doi/10.1021/acs.jcim.0c00025). You can generate one or multiple conformers from a given SMILES. Nevertheless, this is not perfect. What is really perfect in term of 3D conformers ?
I mean, the crystallography structures from which lot of 3D models are based on are only a frozen state without protein that is not truly representative of the ligand - protein 3D interaction in the water.